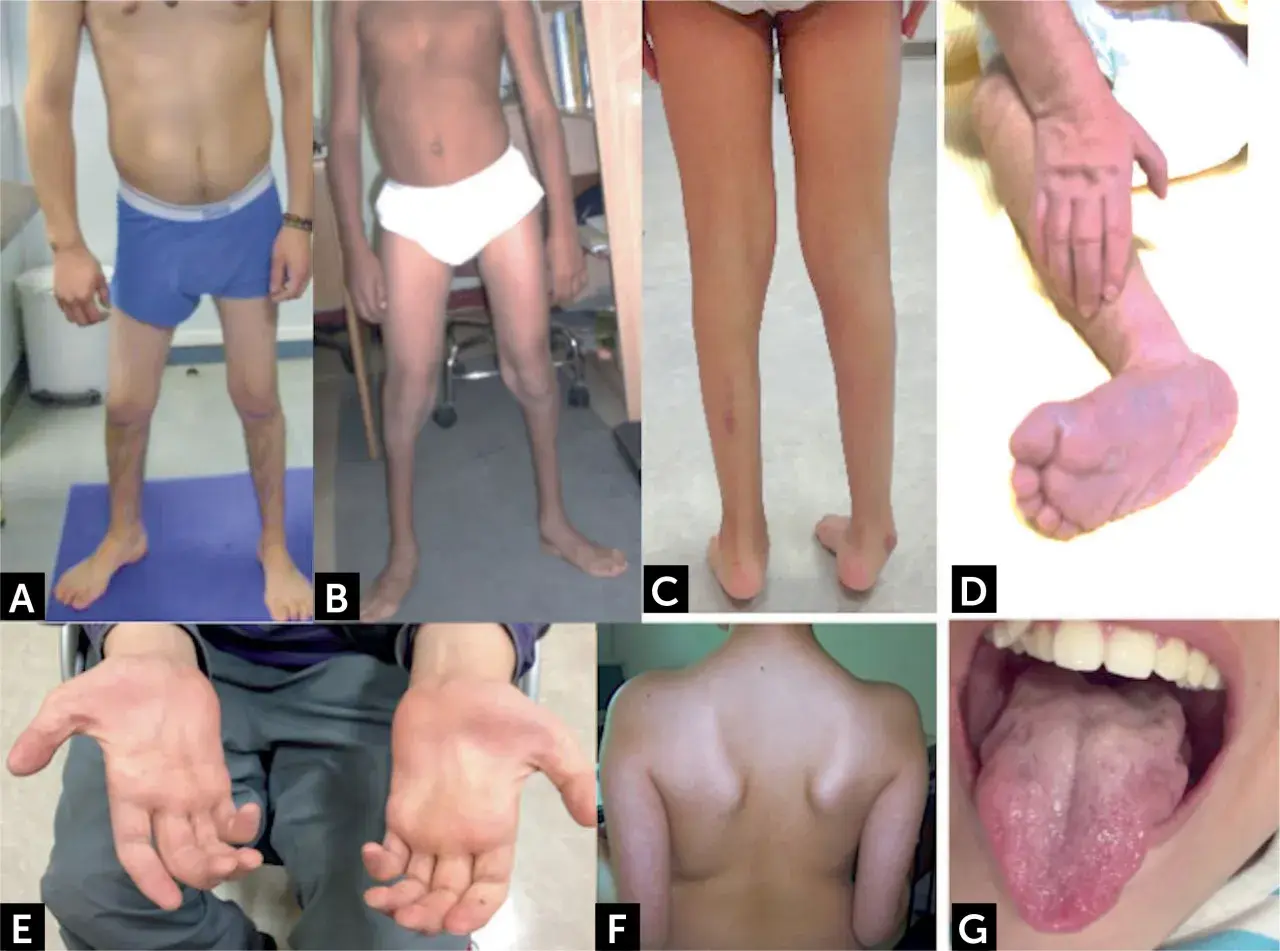
La atrofia muscular espinal (AME) es una enfermedad neuromuscular hereditaria que no afecta solo al músculo: el problema nace en las neuronas motoras de la médula espinal y termina comprometiendo fuerza, postura, respiración y funciones básicas como tragar o mantener la cabeza. En este artículo explico qué ocurre realmente, cómo se detecta, qué tratamientos existen hoy en España y qué puede aportar la fisioterapia para conservar la mayor autonomía posible. También aclaro por qué el diagnóstico temprano cambia el pronóstico de forma muy clara.
Lo esencial para entender el cuadro sin perderse en tecnicismos
- El origen está en las neuronas motoras, no en el músculo como primer problema.
- La debilidad suele ser simétrica y de predominio proximal, y puede afectar la respiración y la deglución.
- La confirmación diagnóstica se hace con genética, y el cribado neonatal acelera mucho el proceso.
- Hoy existen fármacos modificadores, pero el abordaje sigue siendo multidisciplinar.
- La fisioterapia ayuda a movilidad, postura, respiración y prevención de complicaciones.
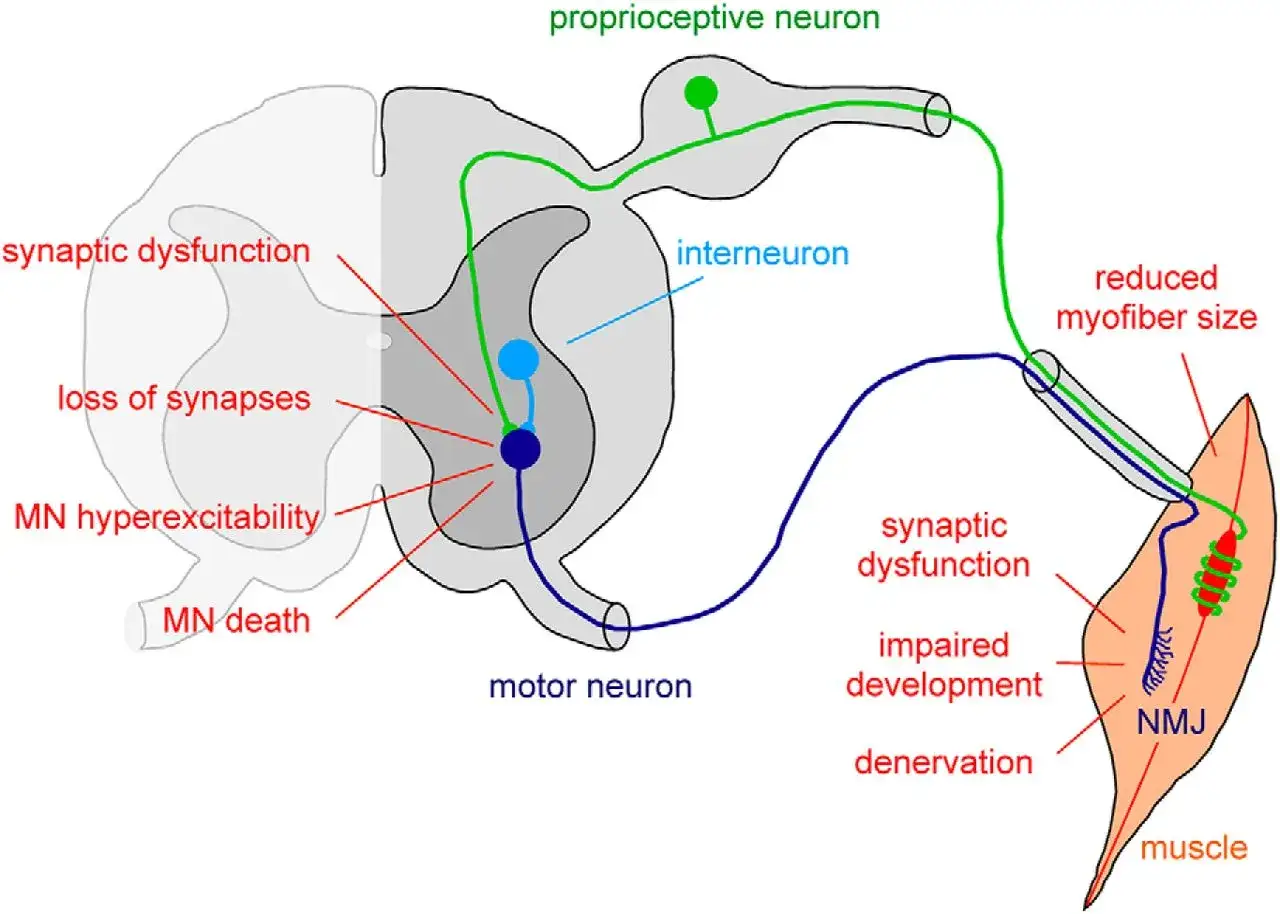
Qué ocurre en las neuronas motoras y por qué el daño progresa
Yo suelo explicarlo así: el músculo no es el origen del problema, sino el receptor del daño. Cuando falla la proteína SMN, las neuronas motoras del asta anterior medular, que son las que llevan la orden del movimiento desde la médula al músculo, se deterioran y dejan de funcionar bien. El resultado es una debilidad progresiva, normalmente simétrica y con predominio proximal, junto con atrofia muscular secundaria.
Esto ayuda a entender por qué muchas personas conservan la sensibilidad y la conciencia intactas, pero notan que les cuesta sostener la cabeza, incorporarse, caminar, toser o tragar. No es una debilidad “cualquiera”: cuanto más tiempo pasa sin intervención, más cuesta recuperar función porque el circuito neuromuscular pierde eficiencia. Con esta base clara, tiene sentido ver cómo cambia la presentación clínica según la edad de inicio.
Cómo se manifiesta según la edad y el tipo
La expresión clínica es muy variable, y por eso conviene pensar en tipos, no en una sola imagen fija. La edad de inicio y la función motora alcanzada orientan mucho la sospecha, aunque no lo explican todo.
| Tipo | Inicio habitual | Rasgos más típicos | Qué suele marcar |
|---|---|---|---|
| Tipo 0 | Antes de nacer | Menor movimiento fetal, debilidad muy precoz | La forma más grave y de inicio más temprano |
| Tipo I | Primeros 6 meses | Hipotonía, poco control cefálico, problemas para comer y respirar | Suele requerir manejo respiratorio y nutricional muy estrecho |
| Tipo II | Entre 6 y 12 meses | Puede sentarse, pero no caminar de forma independiente; escoliosis, temblor fino | La función motora se ve limitada, aunque la evolución puede ser lenta |
| Tipo III | Después de 18 meses, infancia o adolescencia | Camina al principio, luego aparecen caídas, dificultad para escaleras y debilidad proximal | La marcha puede mantenerse durante años, pero con riesgo de pérdida progresiva |
| Tipo IV | Edad adulta | Debilidad leve o moderada, temblores o espasmos, progresión más lenta | Suele ser la forma más leve y de inicio tardío |
La copia del gen SMN2 ayuda a anticipar parte de la gravedad, pero no la determina por completo. En la práctica, yo miro más el conjunto: cuándo empezó la debilidad, qué hitos motores se han perdido, cómo respira la persona y si aparecen problemas de alimentación o escoliosis. Eso marca el siguiente paso, que es confirmar el diagnóstico de forma correcta.
Cómo se confirma el diagnóstico y qué papel tiene el cribado neonatal
La sospecha clínica abre la puerta, pero la confirmación llega con la genética. El estudio busca la alteración del gen SMN1, y cuando el cuadro no encaja del todo, la electromiografía u otras pruebas pueden ayudar, aunque no sustituyen a la confirmación molecular.
El Ministerio de Sanidad señala que el cribado neonatal se realiza con una muestra de sangre del talón entre las 24 y 72 horas de vida y detecta la deleción del exón 7 de SMN1. Si el resultado sale alterado, se hacen pruebas para confirmar el diagnóstico y, si se confirma, se inicia seguimiento precoz en unidades clínicas multidisciplinares. Esa rapidez importa más de lo que parece, porque los tratamientos actuales funcionan mejor cuanto antes se ponen en marcha.Si el inicio es más tardío, la sospecha suele venir por hipotonía, retraso motor, caídas frecuentes, debilidad proximal, tos ineficaz o una dificultad rara para mantener la posición. En ese punto yo no esperaría a “ver si mejora solo”. Pedir valoración neurológica y genética es la decisión correcta.
Qué tratamientos cambian el manejo hoy
Ya no hablamos solo de cuidados de soporte. El objetivo clínico actual es conseguir mejoría o estabilización de la función motora, respiratoria o bulbar, además de una mejor calidad de vida. La elección se individualiza según edad, funcionalidad basal y, en niños, el tipo de afectación y el número de copias de SMN2.
| Tratamiento | Cómo se administra | Para qué se usa | Observación práctica |
|---|---|---|---|
| Nusinersen | Intratecal, mediante punción lumbar | Favorecer la producción de proteína SMN funcional a partir de SMN2 | Requiere visitas hospitalarias y seguimiento especializado |
| Risdiplam | Oral, una vez al día | Modificar el empalme de SMN2 para aumentar proteína funcional | Puede ser útil cuando se busca una opción no invasiva |
| Onasemnogén abeparvovec | Infusión intravenosa única | Aportar una copia funcional de SMN1 | Se indica en pacientes pediátricos seleccionados y requiere evaluación muy cuidadosa |
La parte importante no es solo el nombre del fármaco, sino el contexto. Ninguno borra por completo el daño ya instalado, y por eso la ventana temporal pesa tanto. Cuanto antes se trata a una persona, más margen hay para preservar habilidades. En un caso con síntomas avanzados, la meta suele ser frenar el deterioro y sostener la función que queda, no prometer una recuperación total. Y ahí la rehabilitación deja de ser un complemento para convertirse en una pieza central.
Qué puede aportar la fisioterapia y la rehabilitación diaria
Yo priorizaría cuatro objetivos: mantener movilidad, evitar rigidez y contracturas, cuidar la respiración y sostener una postura útil para comer, sentarse y moverse con menos gasto. En esta enfermedad, la fisioterapia no persigue “hacer trabajar más” al músculo de forma indiscriminada, sino conservar la eficiencia del sistema y retrasar complicaciones que luego cuestan mucho más de corregir.
Lo que suelo priorizar en rehabilitación
- Movilidad articular y estiramientos suaves para frenar contracturas y pérdida de rango.
- Posicionamiento y sedestación para alinear tronco, pelvis y cabeza con menos fatiga.
- Trabajo respiratorio cuando hay tos débil, menor expansión torácica o infecciones repetidas.
- Ayudas técnicas y ortesis para ahorrar energía y proteger articulaciones.
Lee también: ELA - Síntomas iniciales: ¿Qué buscar y cuándo actuar?
Los errores que más veo
- Esperar a que haya una pérdida funcional clara antes de intervenir.
- Confundir reposo con protección, cuando el inmovilismo suele empeorar la rigidez.
- Plantear sesiones demasiado intensas que dejan al paciente exhausto.
- Trabajar sin coordinación con familia, neurólogo, neumólogo y nutrición.
Cuando el plan está bien ajustado, el impacto se nota en cosas concretas, no en grandes discursos: mejor tolerancia a la sedestación, menos rigidez matinal, respiración más eficaz y menos sobrecarga para la familia. Y precisamente por eso conviene saber qué señales indican que el seguimiento se está quedando corto.
Qué señales obligan a revisar el plan y pedir ayuda antes
No esperaría a una crisis respiratoria para mover ficha. Hay signos que merecen revisión rápida, porque suelen anticipar complicaciones que después se vuelven más difíciles de manejar.
- Respiración más rápida, uso visible de la musculatura del cuello o hundimiento costal.
- Tos débil, menos capacidad para expulsar secreciones o infecciones respiratorias repetidas.
- Atragantamientos, comidas muy lentas o pérdida de peso sin una explicación clara.
- Pérdida de habilidades motoras que ya estaban adquiridas.
- Dolor postural, empeoramiento de la escoliosis o dificultad creciente para mantener la sedestación.
- Cansancio excesivo tras esfuerzos muy pequeños o despertar con sensación de mal descanso.
También conviene no aislar el manejo en una sola especialidad. El seguimiento ideal integra neurología, rehabilitación, fisioterapia, neumología, nutrición y, cuando hace falta, ortopedia y apoyo genético. El Ministerio de Sanidad insiste en ese modelo multidisciplinar porque, en la práctica, es el que mejor protege función y evita que cada problema se trate tarde y por separado.
Lo que más conviene recordar cuando se habla de este trastorno neurológico
La idea central es sencilla: el tiempo importa. Diagnóstico genético rápido, tratamiento precoz y rehabilitación bien dirigida cambian mucho más el curso de la enfermedad que la observación pasiva o el reposo prolongado.
Yo me quedo con una regla práctica: si la debilidad aparece pronto, progresa, afecta la respiración o interfiere con la alimentación, no merece esperar. Pedir valoración neurológica y genética, y acompañarla de fisioterapia y seguimiento multidisciplinar, suele ofrecer el mejor margen posible para conservar autonomía y calidad de vida.